


The Painful Truth of Cell Disease
January 31, 2023
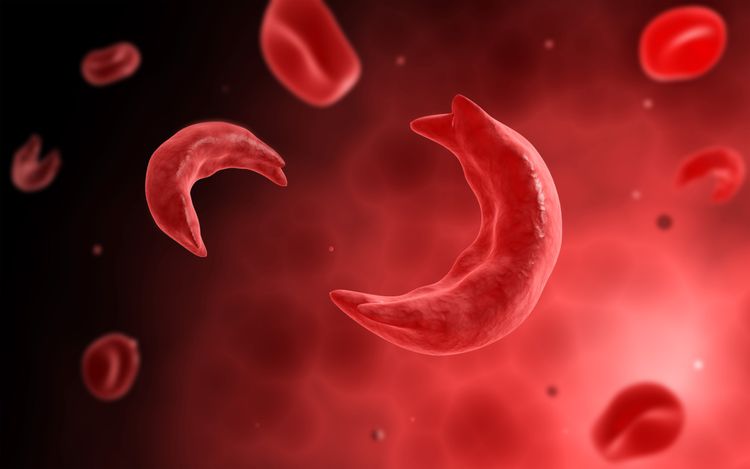
Sickle Cell Disease is a lifelong genetic disorder caused by a genetic mutation. It is characterized by defective hemoglobin, the oxygen-carrying protein in red blood cells. A mutation in the hemoglobin-beta gene, or HBB gene, on chromosome 11 causes an amino acid substitution in which valine replaces glutamic acid. This genetic mutation triggers abnormal hemoglobin S production in the red blood cells. As a result, hemoglobin S subunits stick together, forming rigid and sickle-shaped red blood cells, hence the name Sickle Cell Disease.
In addition to affecting the red blood cell structure, Sickle Cell Disease (SCD) interferes with a cell’s lifespan and ability to deliver oxygen throughout the body. Unlike healthy red blood cells, which live 120 days, defective cells only last 10-20 days, causing a constant shortage of red blood cells. The lack of healthy blood cells results in anemia, a condition in which oxygen delivery and hemoglobin levels decrease. Blockages and blood clots within blood vessels can also cause anemia. Such issues occur due to the atypical structure and stickiness of affected cells.
There are several types of Sickle Cell Disease, all of which are hereditary and dependent on inherited genes. The most common variations include HbSS, HbSC, HbS, HbSD, HbSE, and hbSO. One can only inherit the disease if both copies of their HBB gene, one from each parent, code for abnormal hemoglobin. If both parents have the defect, there is a 25% chance that each child will inherit it. If a child only inherits one copy of the defective gene, there is a 50% chance that they will be a carrier for the trait.
All types of Sickle Cell Disease cause sickle-shaped red blood cells. These defective cells can cause severe episodes of pain, vision issues/loss, delayed growth, jaundice, strokes, dactylitis, organ failure, and more. Those with Sickle Cell Disease are also at a greater risk for infections due to immunodeficiency and abnormal cell structure. These severe symptoms can result in serious medical complications and lead to life expectancy 20-30 years less than those without Sickle Cell Disease.
To date, the only cures for Sickle Cell Disease are bone marrow and stem cell transplants. However, they are costly and high-risk. Instead of surgery, many patients opt for treatments that manage symptoms, like antibiotics, blood transfusions, and pain medications like hydroxyurea. Although such methods have proven successful for some, medical professionals continue to seek new ways to cure and reduce symptoms.
Martin Steinberg, MD, Professor of Medicine at Boston University School of Medicine, said, “What is needed are more drugs that can be taken orally and increase hemoglobin levels.” He believes this will “have a higher likelihood of benefitting populations suffering the most from the disease.”
In addition to the development of oral treatments, medical professionals believe that gene therapy offers the promise of a cure. LentiGlobin is a new gene therapy that collects blood-forming stem cells from a patient’s blood, modifies a copy of the beta-globin gene, and delivers it into the stem cells. The result is the production of healthy red blood cells.
Markus Y. Mapara, MD, Ph.D., Professor of Medicine at Columbia University Vagelos College of Physicians and Surgeons, believes, “You cannot overstate the potential impact of this new therapy.” “This treatment could give people with this disease their life back,” said Mapara. The numerous new developments in gene therapy and Sickle Cell Disease treatments look promising for those affected.
